
Supplementary Figures
Supplementary Table 1. floc and PopPUNK sensitivity and specificity for carbapenem-resistant Klebsiella pneumoniae
| Method | Average Sensitivity | Average Specificity |
|---|---|---|
| floc | 1 | 0.961581971 |
| PopPUNK | 1 | 0.027977687 |
| PopPUNK+PopPIPE (RefDB) | 1 | 0.916195334 |
| PopPUNK+PopPIPE (CustomDB) | 1 | 0.913134983 |
Supplementary Table 2. Further Work Since Poster Creation, PopPUNK+PopPIPE misclustering likely due to custom database
| Method | Average Sensitivity | Average Specificity |
|---|---|---|
| floc | 1 | 0.8901 |
| PopPUNK+PopPIPE (RefDB) | 1 | 0.9035 |
Further Discussion
To determine why PopPUNK is incorrectly separating genetically related isolates from each other (see poster Figure 3), we looked at output data from a PopPUNK+PopPIPE workflow run on our CRAB data when we did not create a custom database. Instead, the PopPUNK reference database for Acinetobacter baumannii was utilized. Table 2 shows the sensitivity and specificity scores for that workflow compared to floc. Results showed that this PopPUNK+PopPIPE RefDB specificity output was slightly increased but relatively equivalent to floc. When the same workflow was run on Klebsiella pneumoniae, we saw that floc had increased specificity when compared to the PopPUNK+PopPIPE RefDB workflow (Table 1).
The explanation as to why the use of our custom database caused PopPUNK to misclassify the isolates is likely found in the way PopPUNK calls clusters. floc estimates pairwise genetic distances between sequences using the sourmash k-mer containment method. floc uses a single k-mer length of 31 and thereby produces a single distance metric for each pair that combines all sources of genomic variation. floc then assigns genomes to clusters using a fixed distance threshold, assigning genomes to a cluster if they fall below the distance threshold or founding a new cluster otherwise. The default threshold utilized (0.03) meant that floc was creating clusters with larger SNP ranges than PopPUNK, but also that we were not seeing any false negatives. In contrast, PopPUNK calculates pairwise distances between sequences across a range of k-mer lengths. By utilizing multiple k-mer lengths, PopPUNK is able to estimate both core and accessory gene distances. PopPUNK’s cluster boundaries are determined by the Bayesian Gaussian mixture model fit, not a static threshold. Therefore, a poor model fit can lead to inaccurate cluster assignment. The isolates we used to create the database were highly clonal, which likely led to a poor model fit. This is likely why we saw the results we did in poster Figure 3.
Although we saw an improvement in PopPUNK performance when using the reference database, it is important for DCLS purposes that the method used performs well for multiple species. Ultimately, we have decided to focus on floc moving forward because of its improved flexibility and output potential.
Supplementary Methods
QC and Genome Assembly
We ran all reads through the CDC/PHoeNIx pipeline, version 2.1.1, to obtain quality control metrics and genome assemblies for each CRAB isolate.
PopPUNK
Database Creation
- Created a custom database of 645 CRAB isolates using poppunk create-db.
- Fit a Bayesian Gaussian Mixture Model to the database using poppunk fit-model bgmm.
Without PopPIPE
- Once the database was created, we queried with another 29 CRAB isolates using poppunk_assign.
- Created visualizations using poppunk_visualise with the microreact flag.
With PopPIPE
- Once the database was created, we ran the PopPIPE workflow on the database.
- Queried the database with another 29 CRAB isolates using poppunk_assign.
- Ran PopPIPE workflow to get subcluster identities for the 29 CRAB isolates.
floc
Database Creation, Query, and Visualization
- Sketched 645 CRAB isolates to create a database. Used k-mer size of 31 and scaled value of 100.
- Queried the database with another 29 CRAB isolates.
- Ran distance output through custom python script (using BioPython Phylo) to obtain neighbor-joining trees.
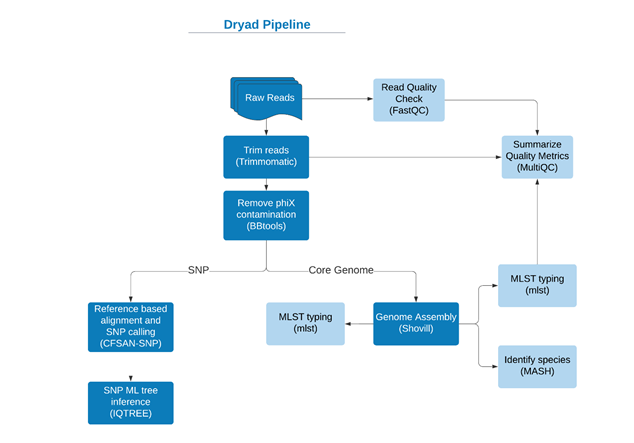
Dryad
Dryad is a DCLS validated reference-based SNP calling pipeline (workflow pictured below). We utilized Dryad on floc cluster 5 (and PopPUNK strain 2, but this was not pictured on poster) to see what the exact SNP range of the floc cluster was compared to the PopPUNK cluster.
Sensitivity and Specificity
Sensitivity was calculated as (true positives) / (true positives + false negatives). Specificity was calculated as (true negatives) / (true negatives + false positives). We calculated sensitivity and specificity for each DCLS cluster, then averaged across all DCLS clusters according to each clustering method (floc or PopPUNK).
- True positives: isolates belonging to the same DCLS cluster assigned to the same floc/PopPUNK cluster.
- False negatives: isolates belonging to the same DCLS cluster assigned to different floc/PopPUNK clusters.
- True negatives: isolates belonging to different DCLS clusters assigned to different floc/PopPUNK clusters.
- False positives: isolates belonging to different DCLS clusters assigned to the same floc/PopPUNK cluster.
References
Lees JA, Harris SR, Tonkin-Hill G, et al. Fast and flexible bacterial genomic epidemiology with PopPUNK. Genome Res. 2019;29(2):304-316. doi:10.1101/gr.241455.118
Johnson J. 2025. floc. https://github.com/DOH-JDJ0303/floc
Hagey JV, Vlachos N, Kent AG, et al. CDCgov/phoenix: v2.2.0. Zenodo. https://doi.org/10.5281/zenodo.8147510
Argimón S, Abudahab K, Goater RJE, et al. Microreact: visualizing and sharing data for genomic epidemiology and phylogeography. Microb Genom. 2016;2(11):e000093. doi:10.1099/mgen.0.000093
Created with BioRender.com
Acknowledgements
This work was supported by the Public Health Laboratory Fellowship Program: an APHL-CDC Initiative. The fellowship assignment was supported by Cooperative Agreement Number NU60OE000104 (CFDA No. 93.322), funded by the Centers for Disease Control and Prevention. This publication’s contents are solely the responsibility of the authors and do not necessarily represent the official views of the Centers for Disease Control and Prevention, the Department of Health and Human Services, or the Association of Public Health Laboratories and member laboratories. The fellowship assignment was 100% funded with federal funds from a federal program of $120,402,978.
We would like to thank the bioinformaticians at the Virginia Division of Consolidated Laboratory Services for mentorship and technical advice - Alexandra Lorentz, PhD; Logan Fink, MS; Rachael St. Jacques, MS; Molly Creighton, MS; and Gretchen Cote, MS. We would also like to thank the Sequencing and Bioinformatics Group at DCLS for their assistance with data processing and sequencing and group manager, Mary Toothman, for keeping everything moving forward. Special thanks also goes to Jared Johnson, PhD of the Washington State Department of Health for technical guidance.